Co-op Documentation
Contents
- 1 Current Tasks
- 1.1 Tasks In Progress
- 1.1.1 Issue Tracker & Bug Fixes
- 1.1.2 Tool Corrections
- 1.1.3 General Page Corrections
- 1.1.4 Refining Interactions widget
- 1.1.5 Gene page alterations
- 1.1.6 Sequence/Transcript/CDS page alterations
- 1.1.7 Variation page alterations
- 1.1.8 Curator Requests
- 1.1.9 Non-compliant Data Checks
- 1.1.10 Interface Corrections
- 1.1.11 Homology Widget
- 1.1.12 Cytoscape plugin
- 1.2 Other Tasks
- 1.1 Tasks In Progress
- 2 Getting Started
- 3 Tools/Resources
- 4 General Info
- 5 General Concepts
- 6 Other Info
Current Tasks
Tasks In Progress
Issue Tracker & Bug Fixes
- Details
- In Progress
- Completed
- #151 - Very useful view for users, from the old site
- CDS JC8.10a
- Gene unc-26
- Ported code from classic site and reformatted to adhere to Model-View-Controller structure
- Adjusted to prevent use of Ace Browser (Configuration). Changed tmp image creation/usage to follow blast/blat method instead.
- Replaced use of image file to display error with standard message display
- Also ported 'Genetic Map' tool
- Added as a sidebar tool for genes and rearrangements as well as a drop-down tool
- Ported and corrected e-PCR tool as well
- #135 - Port the data model CGI to the new site
- Drop-down Index Page
- Gene Model
- unc-26 Schema widget
- Added such that tool should appear wherever tree display appears
- Created basic index page for drop-down
- Implemented by adding case to controller for redirecting schema requests to tree display, and adjusted templates accordingly
- #139 - Need to port (or create anew) a Rearrangment page/display
- #132 - TreeFam IDs on Gene > Homology should link to Treefam, not to WB
- Changed to use "search" in external_urls (Originally using text2link)
- Commit 423c0f
- #127 - Registered Users table broken on MyWormBase for admins
- Changed view to create "row" separately from "obj" (Originally just manipulated obj and pushed onto rows)
- Changed reference used to store role info (Originally wasn't returning role info correctly)
- Commit 3e7c48a
- #82 - evidence in phenotype dataTable
- Changed RNAi evidence retrieval to match alleles & transgenes
- Commit 91da482
- Commit dc5c2b4
- #151 - Very useful view for users, from the old site
Tool Corrections
- Details:
- Tree Display Issues:
- Note: tree display changes postponed because users are primarily dev and curators, primary focus is general correct function for ws230 release
- Clicking opens tree display on separate page
- Create button for opening in new page and keep navigation in the widget (in an iframe)
- Clicking on objects with corresponding pages should actually open those pages with the tree display widget opened and focused
- Allows better navigation using tree display
- Need to test that all types of object can be displayed (particularly links to external images that were using hack method to display)
- Blast/Blat Issues:
- Add Image Map for alignment image that links to the corresponding hit alignment below
- Sidebar architecture
- Tree Display Issues:
- In Progress:
- Tree Display Issues:
- "Can't locate object method "run" via package "WormBase::API::Service::tree" error
- Temporarily removing inclusion of TreeSubs at least allows loading and using
- "Can't locate object method "run" via package "WormBase::API::Service::tree" error
- Tree Display Issues:
- Related/Example Links:
- Completed:
- Blast/Blat Issues:
- mkdir error - doesn't have permissions
Temporarily solved by running server via sudo (probably not viable long term)- chmod -R a+rwx /usr/local/wormbase/shared/tmp/blast_blat
- chmod -R a+rwx /usr/local/wormbase/shared/tmp/media/images/blast_blat
- Corrected UI as discussed in issue 49 on github tracker
- Added version selection and sorted list
- Incorrect linking (genes linking to common names instead of WB IDs. Call to non-existent "common_name" method)
- e.g. beta.wormbase.org/species/c_elegans/gene/lin-1 instead of beta.wormbase.org/species/c_elegans/gene/WBGene00002990
- Was treating the name of the hit as the name of the corresponding sequence, gene, and protein resulting in invalid links
- Was returning links using incorrect hash that tried to mimic what _pack_obj returns
- Changed to fetch sequence object from database, determine actual gene and protein, and return using _pack_obj
- Drop-down navigation list on left of results display doesn't work (trying to switch to another result doesn't do anything)
- Corrected to link to start of display for corresponding result
- Would be nice to have drop-down hover with the screen focus, so that the user doesn't have to click back or scroll to the top to use it again
- Changed sample nucleotide query to include second nucleotide to demonstrate navigation using drop-down
- "About the BLAT algorithm" link is incorrect
- Created template and updated link
- Corrected issue with cross-linking between results with same hits (Alignment View links on results page)
- Corrected Gbrowse [Genome View] link and embedded image for genomic alignments
- Changed to combine hits "HSP(#)" tracks into single track
- Changed retrieval of BLAT databases from being manually defined in hash to being automatically read from the specified database directory in the config (like blast)
- Method for determining BLAT databases in UI list was to manually append "[BLAT]" to genomic BLAST database names
- Changed to read from blat_databases attribute
- Changed method for running from using gfServer and gfClient to use the standalone blat
- Need to handle error when no blat results
- Corrected to inform user and link back to search page
- Test proxy issues
- Gbrowse images
- tmp_image_dir (session binding)
- Handled by Todd
- Corrected reset button on search page to reset fields correctly
- Corrected issue with searching against EST database
- Issue was actually due to the disabling of database drop-down when only one element
- Changed blat to use fasta files in blast directories when searching
- Changed result page navigation drop-down to use onchange instead of onclick
- Disabled linking for CHROMOSOME pages due to loading issues (sequence is too large and blocks acedb for a while)
- mkdir error - doesn't have permissions
- Tree Display Issues:
- Expand/Collapse Nodes and View schema links have invalid links (.../tools/tools/tree/run/...)
- Found issue where Controller passes param that is an array of array refs, but the method treats it as if they have been merged
- Created flatten function "sub flatten {return map { ref($_) eq 'ARRAY' ? flatten(@$_) : $_ } @_}"
- built-in or more efficient method in perl to handle this?
- Embedded links to objects were invalid. Only links to expand/collapse list tags were valid
- Linking was relative to current page which was assumed to be /tools/tree/run
- Changed to always link to tools/tree/run (for ws230 release)
- Changed to open in new tab/window instead of changing the current page
- Model display issues
- Link to object where name had # in name were incorrect
- Fixed by substituting ? for # when generating link
- View schema link broken for Model objects
- Link to object where name had # in name were incorrect
- Blast/Blat Issues:
General Page Corrections
- Details:
- Visiting all minor pages and verifying that all fields display information correctly (checking behaviour for sparsely- & densely-populated)
- Continuing with fatal non-compliance checks for all pages
- Standardization and optimization
- Converting methods to use standard such as _pack_obj/_get_evidence where necessary
- Converting templates to use macros such as build_data_table instead of building manually
- In Progress:
- Feature Page
- Should the SO_term in the overview be removed/changed?
- Gene Class
- If gene class uses other_name (e.g. mig->ppn) shouldn't the genes for both be accessible on the ppn page
- Searching 'mig' gives links to 2 separate pages, where one has mig info and the other has ppn info but the names are opposite
- Both of these pages have ppn as the title when visited
- Searching 'mig' gives links to 2 separate pages, where one has mig info and the other has ppn info but the names are opposite
- If gene class uses other_name (e.g. mig->ppn) shouldn't the genes for both be accessible on the ppn page
- Picture
- Unable to find any pages for this class
- Has API methods, templates, and is defined in wormbase.conf
- Unable to find any pages for this class
- Enabling precaching for widgets/pages with significant load times
- Set "precache true" in wormbase.conf under the widget/page
- Feature Page
- Related/Example Links:
- Completed:
- Analysis Page -> Overview Widget
- Added highlight box back (was originally empty, but now WBID is automatically added)
- Anatomy_function Page -> Associations Widget
- Added references and body parts involved for Anatomy functions
- Reason: No page for anatomy functions and the data should be displayed (also assists in navigation)
- WBbt:0004522
- Added references and body parts involved for Anatomy functions
- Expression_cluster Page -> Clustered_data, Genes, Overview(Remarks) Widgets
- Corrected invalid hash references, and corrected overlap issue for remarks
- http://dev.wormbase.org:8023/species/c_elegans/expression_cluster/[cgc5767]:cluster_88#01243--10
- Corrected invalid hash references, and corrected overlap issue for remarks
- Feature Page
- Corrected improperly displayed (overlapping) remarks in overview
- Corrected array ref issues in associations widget
- Gene Page
- Corrected improper retrieval of phenotypes_not_observed
- Gene Class
- Changed tables in 'Current Genes' and 'Previous Genes' widgets to use build_data_tables
- Consistent, paginated, searchable
- Issue with sparse gene info for previous genes
- Changed tables in 'Current Genes' and 'Previous Genes' widgets to use build_data_tables
- Gene Regulation
- Corrected reference issue for 'molecule regulator' field and modified 'regulates' field to be more clear
- GO_term
- Issue with size: If evidence is shown, GO_term cannot load if it contains > 100 motifs. Otherwise, all GO_terms can load
- Note: largest number of motifs is 880
- Classic site displays human diseases, beta doesn't
- Notes: Tried adding widget to conf (didn't work). Not sure where/how info is being read from for either site
- GO:0018996 (Dev Version)
- GO:0018996 (Classic Version)
- Issue with size: If evidence is shown, GO_term cannot load if it contains > 100 motifs. Otherwise, all GO_terms can load
- Interaction
- Both classic and beta weren't displaying all interactions (possibly due to recent changes to model)
- All information shown was regarding the first(alphabetically) interaction_type (i.e. treating info as scalar instead of array)
- Removed numerous fields and created general summary table showing how everything is related
- Should the Interactors widget be removed/changed? Doesn't seem to provide much useful info
- Should just be replaced by Interactions Cytoscape view
- WBInteraction0004639 (Dev Version)
- WBInteraction0004639 (Classic Version)
- WBInteraction0004639 (Tree Display)
- Restructured table to combine effector, effected, & non-directional into one "Interactors" column
- Both classic and beta weren't displaying all interactions (possibly due to recent changes to model)
- Microarray_results
- Only displayed one gene and one cds (both incorrectly due to tags being arrays but passed to pack_obj as single object)
- Changed to handle as array and use tags2link macro in template
- Added transcript and pseudogene fields
- Added "Experiments" widget containing associated experiments, range, and microarray details
- Aff_Y105C5.DD (Dev Version)
- Only displayed one gene and one cds (both incorrectly due to tags being arrays but passed to pack_obj as single object)
- Molecule
- Corrected affected... fields to display data and formatted into tables
- Created Phenotypes Affected widget and moved relevant fields
- C027492 (Dev Version)
- Operon
- Added Structure widget to display gene information
- Added Location widget with Gene Models and Operon tracks
- CEOP2606 (Dev Version)
- Oligo
- Added In Sequences and PCR products fields to overview
- Strain
- Corrected issue with linking genotype
- Structure_data
- Added overview template and corrected corresponding fields in API
- Added external links widget
- Standardization and optimization
- Combined and cleaned-up API methods for "Gene page->Phenotypes widget->Evidence column of tables"
- Analysis Page -> Overview Widget
Refining Interactions widget
- Details:
- Scale distance based from current gene based on connectedness(number of interactions)
- Shouldn't cluster by gene class (problems arise from classes like "let" where genes aren't necessarily related, so grouping/clustering can be misleading)
- Interesting example where this seems to happen automatically: pptr-1
- Shouldn't cluster by gene class (problems arise from classes like "let" where genes aren't necessarily related, so grouping/clustering can be misleading)
- Scale distance based from current gene based on connectedness(number of interactions)
- Progress:
- Checking cytoscapeweb documentation for how to incorporate this
- Related/Example Links:
- Completed:
- If the number of interactions is too high, only show the minimal table (hide Cytoscape?) and add "Show More"/"Show Neighbouring Interactions"? option
- Otherwise, show all interactions(nearby interactions included) and Cytoscape plugin
- NOTE: This may need to be revisited and optimized/cleaned up
- Looking into BLASTP field in Homology Widget on Protein page that uses similar mechanism
- remove duplication
- instead of toggle href load, load diagram with no nearby in toggle
- create threshold-determining algorithm based on nodes?
- Split interactions into 2 methods, one for those involving current gene and one for nearby interactions
- If local interactions exceeds
400100(after merging duplicates), doesn't load nearby - Combined duplicate edges (duplicate implies matching source node, target node, direction, type, & phenotype)
- Changed table: "Details" column with single link to interaction object -> "Citations" column with list of links to duplicate interactions
- More than 10 results in a cell is now collapsible into "# Results"
- Modified tooltip
- Removed interaction number and link to page (misleading for multiple)
- Added phenotype and number of citations to tooltip
- Added "edge width based on number of citations" feature
- Set 15 as max number of citations; anything higher will be scaled down
- Change colour scheme to follow WormBase's
- Start by looking in main.css
- If the number of interactions is too high, only show the minimal table (hide Cytoscape?) and add "Show More"/"Show Neighbouring Interactions"? option
Gene page alterations
- Details:
- Making corrections to Gene page interface based on curator consultation
- Progress:
- Interactions info needs to be loaded from a separate database to speed up
- Related/Example Links:
- Completed:
- Phenotype:
- Added evidence information
- May need to be modified/re-organized (pending consultation)
- Gene Ontology widget uses similar mechanism
- _get_evidence method in Object.pm
- Added evidence information
- Consultation from curators. Teleconference: 11:30 am EST Wednesday, January 18th, 2012
- Interactions:
- Added edge-colour legend
- Hide no_interaction edges unless that option is selected/filtered for by user
- Phenotype:
- Standardize look and feel of tables (convert to data tables)
- Done changing Phenotypes, Phenotypes_not_observed, and RNAi tables to use build_data_table
- Modified corresponding API methods to return compatible data for build_data_table
- Combined/merged Phenotype and RNAi tables for observed and not_observed (with >10 results collapsible)
- Standardize look and feel of tables (convert to data tables)
- Reagents:
- Drop the word "found" for search results
- Switch search results to actual list of results(collapsible if too many)
- See/learn how this was done for the Expression widget
- implemented using tags2link macro
- Homology:
- restore best blast hits (on the protein object)
- protein domains: alphabetize & restrict to just interpro hits
- Changing evidence in Phenotypes widget to be consistent
- Format: "name/id(paper)" with evidence drop-down when clicked for all types - Alleles, Transgenes,& RNAi(extract info)
- Phenotype:
Sequence/Transcript/CDS page alterations
- Details:
- Making corrections to Sequence, CDS, & Transcript pages based on curator consultation
- CDS
- Sequences Widget
- Show marked-up sequences for corresponding transcripts
- Sequences Widget
- All
- Overview Widget
- Download dialog launched from highlight box: fix scrollbar and rapid-click issues
- Location Widget
- Highlight current on track or at least show gene names
- Overview Widget
- Progress:
- Split CDS and Transcript, now checking that methods do not attempt to access invalid fields
- Related/Example Links:
- JC8.10c (CDS)
- JC8.10c.1 (Transcript)
- yk444b8.5 (Sequence)
- 2RSSE (Sequence w/ Method)
- Completed:
- Consultation from curators. Teleconference: 11:30 am EST Wednesday, January 25th, 2012
- Separating Transcript(+CDS) and Sequence pages
- Separating CDS from Transcript
- Checking Sequence page methods to ensure that only valid fields are accessed
- Comparing with schema
- Removing eval statements
- Region Widget
- Redistribute data and remove widget
- Matching cDNAs moved to Reagents widget
- Moved "Transcripts in this region" field to Sequences widget
- Redistribute data and remove widget
- Origin Widget
- Moved to Overview
- Changed to a curator-only field using curator_block macro
- Reagents Widget
- Add PCR products
- Tags/info should already be in the model
- Add PCR products
- Sequences Widget
- Predicted Exon Structure table
- Removed completely (Although tag exists in schema, it has not been populated for any Sequences, only Transcripts)
Calculate and show exon & intron sizes using "Relative to Itself"Remove "Relative to Superlink"Start & End "Relative to Itself" are useful in some cases (keep)
- Predicted Exon Structure table
- Sequences
- Sequences Widget
- Show orthologs? (for navigation to related info, especially useful for curators)
- Syntenic sequences
- Show corresponding gene in predicted genes and transcriptional units (e.g. unc-26 for yk444b8.5)
- Show orthologs? (for navigation to related info, especially useful for curators)
- Sequences Widget
- Transcript
- Split into CDS and Transcript
- Location Widget
- Show protein domains for CDS
- All
- Overview Widget
- Make origin info available to users (not just curators)
- Show remarks/description associated with Method to make it more meaningful
- e.g. Method for AAA27985.1 is ndb_cds which is not particularly meaningful
- Show affiliated gene(e.g. unc-26 for yk444b8.5)
- Add navigation table (similar to table in sequences widget on gene page, e.g. Matching transcripts/CDS)
- Location Widget
- Hide YAC's, fosmids, & cosmids track
- Overview Widget
Variation page alterations
- Details:
- Making corrections to Variation page interface based on curator consultation
- Change default widgets (Overview, Genetics, Molecular Details, Phenotypes)
- Overview Widget
- Hide Status field if Live (show otherwise e.g. suppressed/dead)
- Remove redundant remarks
- Molecular Details Widget
- Check that the sequence is actually on the + strand?
- "Flanking Sequences" and "Context" should be removed
- Just show marked-up sequence by default (toggle if too large)
- Add colour legend (yellow=flanking, red=deletion/substitution/...)
- Features Affected field -> Predicted CDSs
- Add "Variation" to "Contained in:" to clarify
- Change Predicted CDSs and Clone to data tables
- Location Widget
- Remove contig submission track
- Phenotypes
- Remove remarks and add evidence/citations
- Remove phenotype description or hide by default
- Progress:
- Related/Example Links:
- Completed:
- Completed by Abby
- Consultation from curators. Teleconference: 11:30 am EST Wednesday, February 1st, 2012
Curator Requests
- Details:
- Protein Page (Christian A Grove)
- UNC-26 (Sample Protein Page)
- Overview widget: in the right-hand box, the "Status: history" is not clear. What does this mean? Do we need/want to display this?
- External Links widget: "TREEFAM" links back to the same UNC-26 protein page, as does "WORMPEP". "WP:CE28239" links to the old UNC-26 protein page; is this what we want? Also, both UniProt links are dead/obsolete/outdated
- We should add units to Molecular Weight wherever it is mentioned (as I said in the gene page review). The units should either be "kilodaltons" or "kD"
- History widget: The "JC8.10" under "Predicted gene" links to a WormBase search result for "JC8.10". I think it would be better to direct users to the exact (unc-26) page, rather than having them have to select among many options.
- The red text in the protein schematic for domain names is a bit hard on the eyes. Could we go with a more neutral color: black, grey, blue...?
- Can we include descriptions of the domains depicted in the domain cartoons, or at least provide a link out to the relevant InterPro or PFAM pages?
- Gene Page (Christian A Grove)
- cup-5 (Sample Gene Page)
- Postponed:
- Human Diseases widget:
- The description for the disease is cutoff: "...along the lysosomal pathway, affecting membra" ; is this intentional? Could we maybe add a "..." to the end?
- Phenotype widget
- Cross-platform issues (Chrome & Safari vs Firefox)
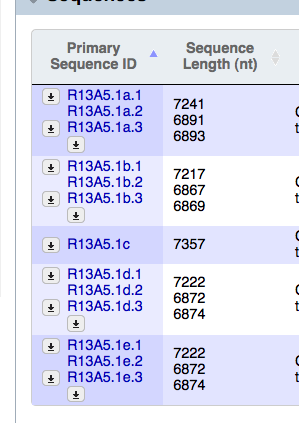
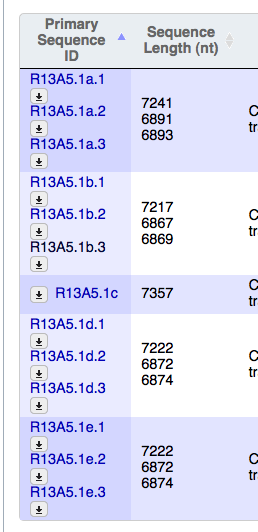
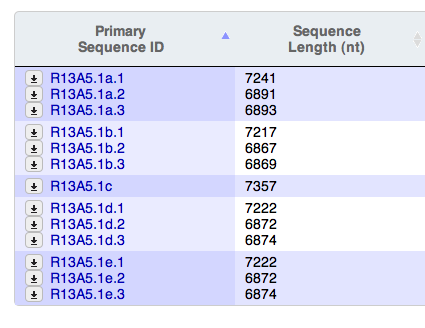
- The table display for "Primary Sequence ID" in the Sequences widget is a little wonky on Firefox. As you can see from the screenshots (attached) as I resize the table in Firefox, that column adjusts strangely, skewing the text, whereas in Safari and Chrome, the table stays nice and neat.
- Human Diseases widget:
- There are a number of unnecessary hyperlinks in the phenotype description details. For example, all remarks, the word "Genotype" after "Phenotype assay:", and the phrase "Uncharacterised_loss_of_function" after "Loss of function:" are all hyperlinked to what appears to be a general WormBase text search result. These should just be plain text.
- One general comment is that I think I would prefer that links out to other pages open a new tab by default, but I may be in the minority with that opinion (or that may have been your intention, but Firefox is not cooperating)
- Protein Page (Christian A Grove)
- Progress:
- Investigating hyperlinks in phenotype description details
- Related/Example Links:
- Completed:
- Gene Page
- External links widget:
- The OMIM link out appears to be malformed. The URL that the link directs me too is:
- http://omim.org/enrty/OMIM:605248 (verbatim, entry is mispelled)
- whereas this is a working link:
- (as I write this I realize that someone from OMIM just e-mailed to point this out)
- Queried ACeDB Database class for databases with URL, URL_Constructor, and Description and added those to the external_urls template
- Need to verify that all links are working and add other links(i.e. databases that didn't match the query)
- Added/updated descriptions (some from ACeDB, some from website's homepage)
- The OMIM link out appears to be malformed. The URL that the link directs me too is:
- Homology widget:
- When I click on the download option for homologous proteins, a window pops up to show Sequence, Isoelectric Point, Molecular Weight, etc. I know this is more a Protein issue, but we should put units for Molecular Weight (e.g. "77.7 kiloDaltons" or "kD" as opposed to just "77.7")
- Genetics widget:
- In both the "Alleles" and the "Polymorphisms & Natural Variants" tables, the right-most column is "Protein effect" and "Location", respectively. Under "Protein effect" is displayed "nonsense" and "missense" (which are OK), but also "intron", which does not make sense. Under "Location" is displayed "intron" and "utr_3" (OK), but also "missense" and "silent" (not OK). I would suggest having a "Protein effect" and a "Location" column in each table and putting only relevant entries in each:
- Protein effect: nonsense, missense, silent, etc.
- Location: intron, exon, utr_3, utr_5, etc.
- In both the "Alleles" and the "Polymorphisms & Natural Variants" tables, the right-most column is "Protein effect" and "Location", respectively. Under "Protein effect" is displayed "nonsense" and "missense" (which are OK), but also "intron", which does not make sense. Under "Location" is displayed "intron" and "utr_3" (OK), but also "missense" and "silent" (not OK). I would suggest having a "Protein effect" and a "Location" column in each table and putting only relevant entries in each:
- Interactions widget:
- The different color nodes and thickness of lines are not explained in the legend. Can we add an explanation somewhere?
- In the "Citations" column is listed all of the WormBase Interaction objects affiliated with this interaction. I would expect to see paper references in a "Citations" column. Could we perhaps put the interactions under a "Interactions" column all the way to the left, and keep the "Citations" column at the right with WBPaper references?
- The Cytoscape network view looks great, but the window for viewing it is too short (vertically) in Firefox, and too tall in Safari and Chrome. Could we find an intermediate height or provide options to resize?
- External links widget:
- Gene Page
Non-compliant Data Checks
- Details:
- Using fatal_non_compliance check and debug option (enable in wormbase_local.conf) to determine if API methods return invalid data
- E.g. returning ACe objects(usually this should be a string) or empty arrays/hashes(should return undef)
- Usually these can be found when fields do not have data to return but give empty structures instead of just undef
- Using fatal_non_compliance check and debug option (enable in wormbase_local.conf) to determine if API methods return invalid data
- Progress:
- Correcting these as they're encountered
- Completed:
- Methods showing errors on the following pages were corrected:
- Variation, Person, Laboratory, Gene Class, Life Stages, Phenotype, Protein, Strain
- Methods showing errors on the following pages were corrected:
Interface Corrections
- Details:
- Look into rearranging the elements in widgets so that they do not appear strange/distorted when switched from one- to two-column views or using smaller displays
- Will require modifying the templates
Homology Widget
- Needs to be modified; Contains too much information that shouldn't necessarily be grouped this way
- May need to move orthologs to separate widget
Cytoscape plugin
- Incorporate button/option to open pop-up for larger view (e.g. in case user has small screen or is using 2-column layout)
- Also, add plugin to interaction page (may need to wait until interactions are merged)
Other Tasks
Additional tasks that require further investigation
- Loading/processing large amounts of data
- Details:
- Likely approach will be to change the database used from ACeDB to NoSQL/CouchDB
- Sample Issue
- Progress:
- Ace2Couch Scripts for migrating data
- AceCouch Perl API
- Details:
Getting Started
Members/Contacts
- Abigail Cabunoc - abigail.cabunoc@oicr.on.ca
- Todd Harris - todd@wormbase.org
- Lincoln Stein - lincoln.stein@gmail.com
- Quang Trinh
Meetings
- WormBase OICR Developers Teleconference
- Mondays, 3:00PM
- Phone-in 1-800-747-5150 id: 6738514
- WormBase OICR Developers Teleconference with Lincoln
- Wednesdays, 4:00PM EST
- Lincoln's office
- Agenda and Minutes
- WormBase International Group (HelpDesk) Teleconference
- Every other Thursday, 11:30AM
- Lincoln's Office
- Alternatively: 1-866-528-2256
- Access Code: 714646
- Conference calls
- Group Meeting
- Fridays, 3:00PM
- HL31 Conference Room/TBA
Tools/Resources
The following are some of the tools with which many major aspects of WormBase are developed. You should familiarize yourself with them through documentation and examples.
Perl
There is significant documentation on getting started with Perl. One starting point is PerlMonks For information regarding the use of ACeDB in conjuction with Perl (e.g. retrieving data), check AcePerl Documentation. The Ace::Object section contains most of the information related to interacting with ACeDB objects.
Catalyst
Catalyst is the web development framework used to develop WormBase. To get started, read and try examples from
- The Definitive Guide to Catalyst (should be available on bookshelf)
- CPAN - About Catalyst
Git
Git is a version control system used for collaboration and backup in the development process. One starting point is the progit tutorial
- WormBase repository located at https://github.com/organizations/WormBase
- Common commands
- status, add, checkout, commit, push, pull, log, fetch, merge
- Usage example:
Given that we have modified two files a.txt and b.txt but do not wish to keep the changes made to b.txt
git status
git checkout b.txt
git add a.txt
git commit -m “Added change1 and change2 to a.txt”
git push
If we have did not have the most recent version, then we will run into an issue when trying to push. In this case we can:
git fetch
git merge
git push
Note: pull is similar to using fetch + merge
Browser (Debugging)
Many browsers provide useful tools to developers that can be used for debugging
Chrome
- Tools->Developer Tools/JavaScript Console
Firefox
- Firefox->Web Developer->Web Console/Error Console
Other
Some other tools that you should be aware of but may not be required to know/interact with include:
- JavaScript
- ACeDB
- Usage:
- Navigate to /usr/local/wormbase/acedb/bin and run: "./tace ../wormbase"
- For more help, try the tace tutorial
- Usage:
- MySQL
- Xapian
- GFF
- Cytoscape Web
- Cytoscape Tutorial
- Plugin generally used for pathway analysis
- This plugin is used in the Interactions widget on the Gene page ([WormBase dir]/root/templates/classes/gene/interactions.tt2)
- Installation directory (for updating): [WormBase dir]/root/js/jquery/plugins/
General Info
- Unix and Cluster training by Quang (OICR login required)
General Concepts
Starting the server
- In a terminal, connect to wb-dev (via ssh) and navigate to the root of your dev site (where you checked-out the git repository e.g. /usr/local/wormbase/website/[user]/)
- Run the startup script: "./script/wormbase_server.pl -p XXXX" where XXXX is a port number
- Typical options are -d for debugging and -r to auto-restart when changes to the code are detected
- Output can be redirected into a log file ">>logs/server_log.log 2>&1" (redirects both output and errors into server_log.log)
- Therefore a full command could be "./script/wormbase_server.pl -d -r -p 8023 >>logs/server_log.log 2>&1"
- Logs can be monitored in real-time by running "tail -f logs/server_log.log" or "tail -f logs/wb-dev-catalyst.log" in a separate terminal
- Check Unix and Cluster training by Quang for useful Unix commands (OICR login required)
Widget Data Loading

REST Controller:
- Catches internal url (/rest/widget/...)
- Determines the class and widget from the url
- From class and widget, determines which fields are required from the configuration file (wormbase.conf)
- API methods request data from appropriate databases, process/format/package the data, and then return it(/lib/WormBase/API/Object/[Class].pm)
- Sends data to the template to be used in rendering the widget (/root/templates/classes/[Class]/[Widget].tt2)
Tool Data Loading

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Typical flow of information:
- Request information such as the url and parameters are passed from the browser to the server
- Tools controller in server processes requests to /tools/... and calls corresponding API method
- Determines tool and action from url (e.g. /tools/tree/run... => tool=tree, action=run)
- Note: schema and gmap tools have similar function to other tools, tree and epic respectively
- Any calls to these are translated to calls to the corresponding tool (e.g. schema->tree or gmap->epic)
- Processes special parameters such as inline (for embedding in widget)
- Calls the API method corresponding to the action (e.g. "run" in tree.pm)
- Determines tool and action from url (e.g. /tools/tree/run... => tool=tree, action=run)
- API method processes information passed from Controller and returns processed/packaged info to Tools Controller
- Loads data passed from Controller(e.g. "my $name = $params->{'name'};" loads the name)
- Loads appropriate data from databases (e.g. ACe object corresponding to the given 'name' & 'class' params)
- Performs subsequent processing on data (e.g. blast tool runs blast with the given params)
- Formats/packages data (e.g. commonly used method is _pack_obj which stores appropriate info for creating links for the interface)
- Note: it is important to not return raw data (primarily ACe objects) to the interface. Two possible fixes are _pack_obj($object) or "$object" (stringify)
- Note: it is also important to return "undef" instead of null or empty arrays/hashes
- Tools Controller passes API results to View (Template)
- View (Template) arranges/outputs the data for display
- Uses various methods/macros
- tag2link to convert a hash generated using _pack_obj into a link
- build_data_table to generate a formatted data table
- Note: common/useful macros can be found in /root/templates/config/main and /root/templates/shared/page_elements.tt2
- Uses various methods/macros
Other Info
Useful Macros when editing templates can be found in:
- /root/templates/config/main
- e.g. tags2link
- /root/templates/shared/page_elements.tt2
- e.g. build_data_table
Useful files for debugging include:
- /logs/wb-dev-catalyst.log
Other:
- If the port you have been using appears to be busy but the server is not running,
ps -aux|grep -XXXX kill -9 ID
where XXXX is the port number(e.g. 8023) and ID is the process id that is using the port(e.g. 10361) NOTE: Do not kill other users' processes if you are using a shared dev machine (e.g. wb-dev)
- To dump data in API methods include Data::Dumper
use Data::Dumper ... warn(Dumper(\@data));
- Sometimes firewall blocks certain ports, so you may be unable to connect to your dev server on the wb-dev machine through the browser
ssh -L 8080:localhost:XXXX wb-dev.oicr.on.ca ./wormbase_server.pl -p XXXX -d -r
Then go to localhost:8080 in browser